近日,我院新能源材料与传感器件团队刘震老师与研究生作为第一作者在能源期刊Nano Energy(IF=17.8)连续发表两篇高水平论文。这两项成果重点突出氧策略作为调控异质结构界面化学环境与电子结构的重要方法,并结合贵金属等离激元效应的光学特性促进电荷转移,提高催化剂的电催化析氢性能,实现能源的高效利用。主要研究内容包括:
(1)氧调控Ru纳米团簇在Co3O4多孔纳米线界面电荷分布增强电催化析氢反应。制备了多孔Co3O4纳米线负载钌纳米团簇纳米催化剂 (Ru/Co3O4 NWs),利用球差校正电子显微镜和扩展的X射线吸收精细结构等方法证实了Ru-O键的存在。实验和理论计算研究氧在调控Ru原子的配位环境及电子结构中的作用。Ru/Co3O4 NWs在碱性介质中的HER性能优于20 wt.% PtC商业催化剂。通过Ru纳米团簇在泡沫Co电极 (Ru/CF)上的阳极氧化掺氧,进一步验证了氧策略提高Ru 产氢活性的普适性与有效性。
(2)通过Au-O键强耦合实现等离激元增强析氢反应动力学。采用光还原法制备了Au负载MoO2异质结构纳米片(Au-MoO2)。X射线吸收精细结构表征Au-MoO2界面结构,KPFM测量和有限元仿真研究等离激元光学性能。研究Au-MoO2在等离激元促进情况下的HER性能。通过对贵金属-金属氧化物界面活性位点分布和电荷转移机制的深入研究,阐明Au-O键的耦合作用对界面电子结构及氢吸附能的影响。
氧调控Co3O4多孔纳米线上Ru纳米团簇的电荷分布及增强析氢反应

文章信息
第一作者:刘震、曾丽丽
通讯作者:刘宏教授、周伟家教授
通讯单位:BWIN必赢官网,山东大学
DOI: 10.1016/j.nanoen.2021.105940
文章简介
近日,7321com必赢周伟家教授和刘宏教授团队等人报道了一种氧策略调控钌原子在催化剂载体上的配位环境及电荷分布并增强析氢反应的方法。该研究通过阳离子置换和控制还原合成制备了多孔Co3O4纳米线负载钌纳米团簇纳米催化剂 (Ru/Co3O4 NWs),利用球差校正电子显微镜和扩展的X射线吸收精细结构等方法证实了Ru-O键的存在。与Co纳米线负载钌纳米团簇(Ru/Co NWs)相比,Ru/Co3O4 NWs在碱性介质中具有更好的HER性能(电流密度为10mAcm−2时过电位为30.96 mV,电化学面积为4.31 mF cm−2),而且比20 wt.% PtC商业催化剂具有更好的稳定性。理论计算还表明,由于O的键合使Ru原子的电荷密度发生重新分布,Ru/Co3O4 NWs具有更小|ΔGH*|的值。最后,通过Ru纳米团簇在泡沫Co电极 (Ru/CF)上的阳极氧化掺氧,进一步验证了氧策略提高Ru 产氢活性的普适性与有效性。这项工作为探索高效Ru基HER催化剂制备策略提供了一个有价值的方向,具有广泛的工业应用前景。相关成果以“Charge Redistribution of Ru Nanoclusters on Co3O4 Porous Nanowire via the Oxygen Regulation for Enhanced Hydrogen Evolution Reaction”为题,发表在国际著名期刊Nano Energy上。刘震老师和研究生曾丽丽为本文共同第一作者。
背景介绍
水电解制氢是一种绿色制氢技术,高效催化剂的应用可以显著降低能耗。钌基催化剂在析氢反应中得到了广泛的研究,在酸性和碱性环境中,钌基催化剂均能表现出较高的催化性能。理想催化剂的催化位点对催化反应的中间体应具有适中的吸附强度,研究发现当施加低于0.04 V (vs. RHE)的电势时,钌基电催化剂表面几个原子层可以被还原为金属钌,而在Ru上H*的吸附能太强,导致析氢活性被抑制。因此,合理的调节催化剂表面钌原子的吸附能对提高HER催化性能具有重要意义。通过阴离子的掺杂可以调控中心原子的电荷分布及微观电子结构,降低中间体的吸附能从而提高产氢活性,如氮、碳、硫和磷等元素。通过氧掺杂可以改变中心原子的配位环境,通过金属-氧键影响其电子结构及电荷分布调控其H*中间体的对吸附能。利用氧掺杂策略调控Ru的电荷密度分布及其HER催化性能,现阶段还较少报道。很多研究工作将精力集中在寻找Ru的各种新型载体材料,对载体中氧元素对配位环境的影响关注较少,对氧的协同作用和活性位点分布也缺乏深入地了解。因此,深入探讨Ru基催化剂在载体形成过程中发生的配位环境及电子结构变化,依据催化位点的相对优势设计更高效的Ru基催化剂,并将这一经济有效的策略应用于工业应用领域会具有良好的前景。
本文要点
本文为了探讨氧的调控作用,首先利用载体的晶格氧引入氧元素来调节Ru的配位环境形成Ru-O键合。采用阳离子置换和控制还原法分别制备了多孔Co3O4纳米线 (Ru/Co3O4 NWs)和多孔Co纳米线负载的Ru纳米团簇 (Ru/Co NWs)。
1. Ru/Co3O4 NWs结构表征
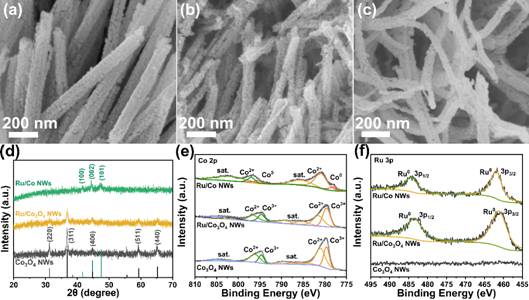
图1 Co3O4 NWs,Ru/Co3O4 NWs和Ru/Co NWs材料形貌表征和结构分析
FESEM图像所示。可以观察到Co3O4 NWs具有直径为50~100 nm、长度为许多微米的多孔纳米线形态。Ru/Co3O4和Ru/Co NWs的多孔纳米线形貌保持不变 (图1b和1c)。图1d为合成过程中XRD图谱的变化, Co3O4 NWs在引入Ru后经阳离子交换和125℃还原后晶面没有发生变化。图1f中XPS分析显示Ru/Co3O4 NWs(低温还原)和Ru/Co NWs(高温还原)均检测到Ru(0)的3p峰值分别位于461.5 eV和483.75 eV,证明在较低还原温度(125℃)下,氧化钌首先被还原为金属钌,而氧化钴保持稳定,在350℃下氧化钌和氧化钴均还原为金属态。

图2 Ru/Co3O4 NWs的TEM图像(a)、HRTEM图像(b)、元素mapping(c)和HAADF-STEM图像(d, e)。
Ru/Co3O4 NWs(图2a)的TEM图像显示出类似的直径为50 nm的多孔纳米线形貌,这与FESEM结果一致。图2b中HRTEM结果与图1d中的XRD结果也一致中 0.46和0.28 nm的晶格条纹归属于Co3O4的(111)和(220)晶面,Ru/Co3O4 NW由Co、O和Ru元素组成(图2c),因此,Ru/Co3O4 NWs中钴元素主要以氧化态形式存在。然而,在HRTEM图像中并没有发现Ru的晶格特征,所以采用球差校正扫描透射电子显微镜(ASSTEM)获得Ru/Co3O4 NWs的HAADF图像,如图2d和2e所示,在Ru/Co3O4 NWs中,Ru元素主要以纳米团簇的形式存在,电感耦合等离子体发射光谱(ICP-OES)结果也证实了Ru/Co3O4 NWs中Ru含量约为5.45%。
2. Ru/Co3O4 NWs原子配位环境表征
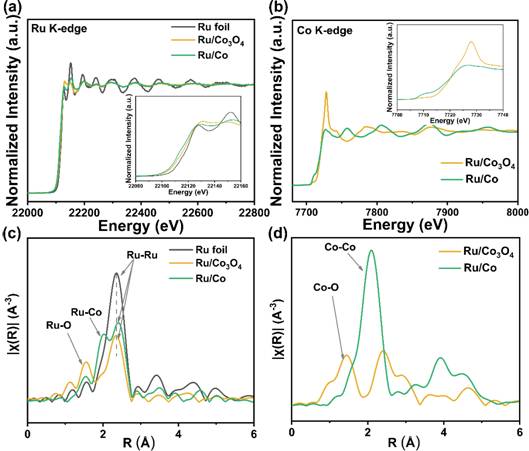
图3 Ru箔、Ru/Co3O4 NWs 和Ru/Co NWs的X射线近边吸收结构谱和扩展X射线吸收精细结构
x射线吸收近边结构(XANES)光谱显示在Ru/Co3O4 NWs 和 Ru/Co NWs 中Ru K吸收边的强度低于Ru箔,这表明Ru的配位环境中可能存在与Co或O的成键。图3c中的扩展x射线吸收精细结构(EXAFS)显示在Ru/Co3O4 NWs中同时存在Ru-Ru和Ru-O键,而不存在Ru-Co键,Ru/Co NWs有两个突出的峰,分别对应于Ru-Co键和Ru-Ru键。这些结果证实了Ru在Co3O4 NWs和Co NWs载体上主要以纳米团簇形式存在,特别是在Co3O4 NWs上通过Ru-O-Co与Co3O4相结合。Ru的配位环境会显著影响可以影响的电子结构和电荷分布,这在催化活性方面将起着重要的作用。
3. Ru/Co3O4 NWs电催化析氢性能
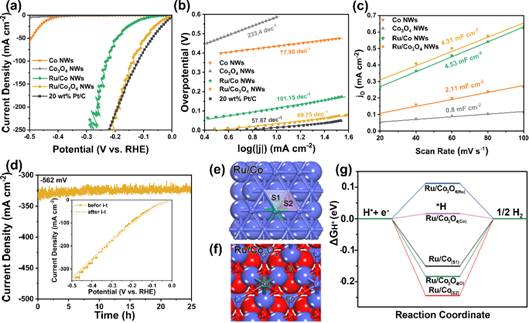
图4 HER的性能电化学表征及DFT计算|ΔGH*|结果
考察了Ru/Co3O4 NWs和对照样品在1.0 M KOH中对HER的电催化活性。图4a为HER在Co NWs,Co3O4 NWs,Ru/Co NWs、Ru/Co3O4 NWs和20 wt.% Pt/C上的极化曲线。Ru/Co3O4 NWs在10 mA cm−2的过电位为30.98 mV,远低于Ru/Co NWs的过电位(113.29 mV),催化性能接近20 wt. % Pt/C。图4 b 中Tafel斜率显示Ru/Co3O4 NWs的HER决速步骤是电化学解吸过程(Volmer-Heyrovsky机制),电化学表面积结果也表明其催化性能取决于材料的本征催化活性,而不是载体差异的影响。在过电位为562 mV(图4d)的情况下,通过i-t测试来评估Ru/Co3O4 NWs的催化稳定性(图4d),在25 h的测试过程中,电流密度保持稳定性,经过i-t测试后,多孔纳米线结构仍然存在。DFT计算结果显示,Ru/Co3O4中Ru、Co和O的|ΔGH*|分别为0.11、0.02和−0.18 eV,并且当Ru/Co中引入O后,Ru和Co位|ΔGH*|值也变小,并接近于零,表明HER活性增强(如图4f)。另一方面,见文章中附件图S12电荷密度差分所示,Co3O4晶格面上的Ru原子周围存在缺电子区域(绿色),Co原子周围存在电荷积累区(蓝色),表明电荷从Ru原子向O原子发生了转移。因此,氧的配位作用可以调控Ru的电荷分布,并通过Ru/Co3O4中Ru-O-Co键的耦合作用提高材料的HER本征活性。
4. O-Ru/CF电极工业化应用评价
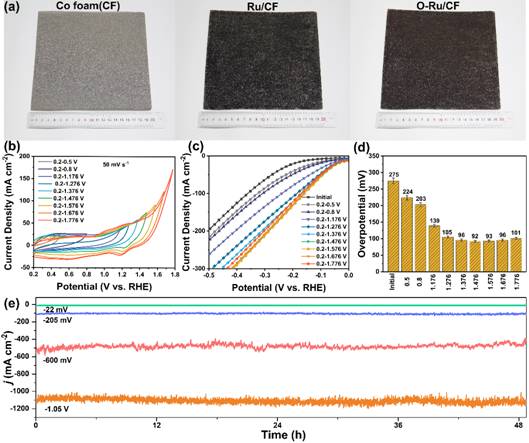
图5 CF、Ru/CF和O-Ru/CF电极的照片及O-Ru/CF的HER性能电化学表征
最终,进一步验证了氧调控策略在工业领域的应用前景,制备了面积为20 cm×20 cm Ru纳米颗粒的Co泡沫电极(Ru/CF),并采用阳极氧化实现了氧掺杂工艺提高HER性能 (O-Ru/CF)。如图5c, 5d所示。极化曲线的响应表明,O-Ru/CF的HER活性与最大氧化电压有相关性,随着最大氧化电压的增加,过电位逐渐减小,形成澡盆曲线。在1.376 ~ 1.676 V的氧化电压范围内,过电位最小。以+0.2~+1.476 V vs. RHE阳极氧化合成的O-Ru/CF电极为例,在10 mA cm−2、100 mA cm−2、500 mA cm−2和1000 mA cm−2电流密度下48小时内均具有较强的HER催化活性性,i-t测试中电流密度保持稳定,衰减可以忽略不,以上结果证明氧调控策略在工业应用领域的通用性前景。
总结与展望
1. 通过阳离子置换和控制还原法成功制备了Co3O4多孔纳米线负载Ru纳米团簇催化剂(Ru/Co3O4 NWs)。
2. 采用球差校正的透射电子显微镜和扩展X射线吸收精细结构等表征技术,揭示了Ru的配位环境及Ru-O键的存在,理论计算进一步证明配位氧可以促进Co基体上Ru的电荷重新分布,从而显著降低Ru的|ΔGH*|值,增强HER活性。
3. 这种具有通用性特点的氧调控策略具有良好的经济效益和工业应用前景。
这项工作进一步揭示了Ru基HER催化剂中氧调节的作用。
文章链接
Zhen Liu#, Lili Zeng#, Jiayuan Yu, Linjing Yang, Jing Zhang, Xiaoli Zhang, Feng Han, Lili Zhao, Xiao Li, Hong Liu*, Weijia Zhou*, Charge Redistribution of Ru Nanoclusters on Co3O4 Porous Nanowire via the Oxygen Regulation for Enhanced Hydrogen Evolution Reaction, Nano Energy, 2021, https://doi.org/10.1016/j.nanoen.2021.105940.
第一作者介绍
 刘震,山东大学博士,现7321com必赢讲师、博士后。主要研究方向为敏感光电材料在能源催化及分析传感器件的应用。在Nanno energy,Nano-Micro Letters,Biosensors and Bioelectronic,Sci. Rep.等发表多篇论文。主持山东省自然科学青年基金,参与重大科学仪器专项,重大基础研究基金子课题等项目。
刘震,山东大学博士,现7321com必赢讲师、博士后。主要研究方向为敏感光电材料在能源催化及分析传感器件的应用。在Nanno energy,Nano-Micro Letters,Biosensors and Bioelectronic,Sci. Rep.等发表多篇论文。主持山东省自然科学青年基金,参与重大科学仪器专项,重大基础研究基金子课题等项目。
 曾丽丽,2017年毕业于福州大学,获学士学位。同年进入华南理工大学新能源研究所周伟家教授领导的能源催化材料组,现为7321com必赢联合培养。主要研究方向为能量转换和存储的纳米材料设计合成及应用,包括为电催化,光催化水分解,CO2还原方向和储能器件。
曾丽丽,2017年毕业于福州大学,获学士学位。同年进入华南理工大学新能源研究所周伟家教授领导的能源催化材料组,现为7321com必赢联合培养。主要研究方向为能量转换和存储的纳米材料设计合成及应用,包括为电催化,光催化水分解,CO2还原方向和储能器件。
通过Au-MoO2异质结构的Au-O键强耦合实现等离激元增强析氢反应动力学

文章信息
第一作者:刘震、姜迪
通讯作者:刘宏教授、周伟家教授
通讯单位:BWIN必赢官网,山东大学
DOI: 10.1016/j.nanoen.2021.106302
文章简介
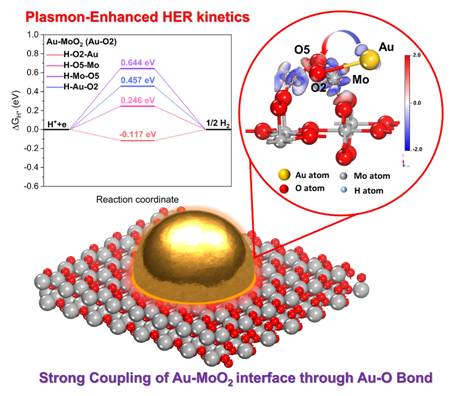
近日,7321com必赢刘宏教授和周伟家教授团队等人报道了一种通过Au-MoO2异质结构的Au-O键强耦合实现等离激元增强析氢反应动力学的方法。通过光还原法制备了负载 Au 的 MoO2 纳米片 (Au-MoO2) 的异质结构。 EXAFS 分析证实了Au-MoO2界面通过Au-O键的实现强耦合相互作用。通过KPFM和FEM模拟在Au-MoO2界面结构区域观察到较强的局部电磁场。得益于Au纳米结构的表面等离激元效应,在光激发下Au-MoO2纳米片的HER活性显着增加,过电势减小(Δη)= 176 mV,并且Tafel斜率也显著降低,表明电极反应动力学过程发生转变。 DFT计算证实了Au-O键中Op轨道的显著提升。本研究通过对Au-O键电荷转移机理及活性位点分布的研究,深入了解表面等离激元促进HER反应的内在机制。相关成果以“ Plasmon-Enhanced Hydrogen Evolution Reaction Kinetics through the Strong Coupling of Au-O Bond on Au-MoO2 Heterostructure Nanosheets ”为题,发表在国际著名期刊Nano Energy上。刘震老师和研究生姜迪为本文共同第一作者。
背景介绍
水电解制氢是一种绿色制氢技术,高效催化剂的应用可以显著降低能耗。制氢是实现氢电交换有效利用的关键因素,也是解决能源问题的有效途径。过渡金属化合物被认为是有效和稳定的非贵金属析氢催化剂之一。过渡金属化合物的电催化性能仍然受到限制,首先,大多数非金属元素催化剂的电导率差。其次,活性位点通常位于表面和表面下,结晶材料不容易暴露。为了解决这些问题,大多数研究集中在提高电荷转移效率以及如何获得更多的活性位点上。许多研究表明,可以通过在两种不同的电催化剂组分之间引入界面来改善催化性能,该界面还可以稳定活性表面上的催化位点并产生协同效应。
贵金属纳米粒子可作为助催化剂,特别是具有表面等离激元效应的贵金属纳米粒子,是提高催化性能的有效途径之一。一方面,等离激元纳米结构可以在较宽的光谱范围内扩展光催化剂的光捕获,而高能等离激元载流子可以与主催化剂的电子-空穴对起到强大的互补作用。另一方面,等离激元引起的电磁场增强将显着提高吸附分子的激发概率,有利于分子活性穿过能垒。金纳米粒子具有优异的表面等离激元性能,已被用作许多光电催化反应。而且,通过施加势场,金属的费米能级与半导体的导带之间的退化将帮助载流子克服肖特基势垒。表面等离激元可在电催化反应中发挥更多的增强作用。
界面处具有很强相互作用的金属氧化物-金属异质结构材料在许多等离激元增强的催化反应中表现出优异的性能,例如水分解,CO2还原,固氮。单斜晶超薄MoO2纳米片具有所需的低电阻率,高表面积,高密度表面位点,这使其成为助催化剂的极好载体。由于d带结构中的富电子态,MoO2纳米片在可见光附近的红外区域也具有可调谐的等离子体共振。MoO2纳米片基质可以实现与负载的等离激元金属的协同作用,从而提高催化活性,但是对协同效应如何发挥作用以及活动位点的如何分布缺少更深入的了解。在本文中,我们采用光还原法构建了具有强耦合特征的Au-MoO2纳米片异质结构催化剂。Au-MoO2在中性介质中表现出优异的等离激元增强的HER活性,与非光照条件相比,实现了过电势的显着降低和电流密度的提高,通过实验和理论计算研究其增强机理。
本文要点
1. Au纳米颗粒负载MoO2纳米片结构表征
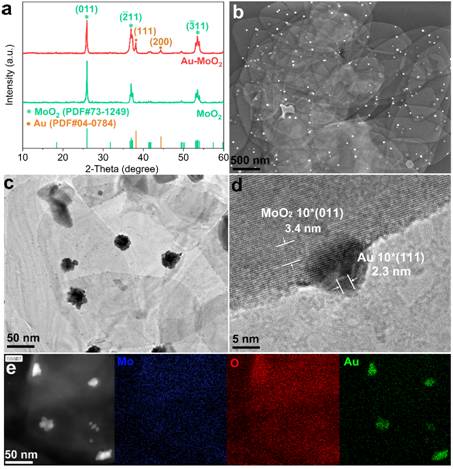
图1 (a)MoO2和Au-MoO2纳米片的XRD图;(b)FESEM图像;(c,d)TEM和HRTEM图像;(e)HAADF-STEM图像以及Au-MoO2的Mo,O,Au成像。
图1a中的XRD图谱显示,MoO2纳米片的特征峰出现在26.0°,36.9°,41.8°,49.4°,53.5°和53.9°,分别属于单斜晶相的(011),(020),(210),(-122)和(-311)晶面(PDF#73-1249,a = 5.611 Å,b = 4.856 Å,c = 5.628 Å,P21 / c(14))。负载金纳米颗粒后,纳米片结构没有显着变化(图1b)。除MoO2特征峰外,在38.1°和44.3°处观察到Au的特征XRD峰,属于立方相的(111)和(200)平面(PDF#04-0784,a = 4.078 Å,b = 4.078 Å ,c = 4.078 Å,Fm3m(255))。如图1b和4-1c所示,在MoO2纳米片上观察到大量的AuNPs,尺寸为10-50 nm。图1d中的HRTEM显示出在AuNPs沿MoO2晶格生长,异质结构的结合区域也具有单晶结构,其中0.34 nm的晶格条纹为MoO2的(011)平面,0.23 nm的晶格条纹为Au的(111)晶面。通过HAADF-STEM和元素成像进一步确定了Au-MoO2的组成,层状结构主要由Mo,O组成, AuNPs生长在纳米片的表面和边缘上(图1e)。 AuNPs和MoO2纳米片之间的牢固结合将使活性表面上的催化位点稳定,并减少电荷转移中的肖特基势垒。具有表面缺陷的MoO2纳米片会使反应物具有特殊的吸附性能和催化活性。
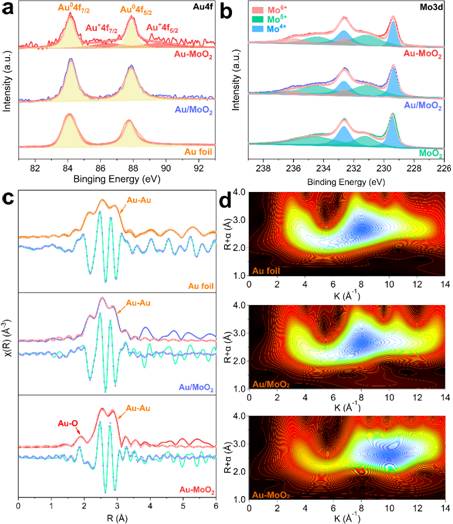
图2 MoO2,Au/MoO2和Au-MoO2纳米片的XPS和EXAFS分析:(a)Au4f XPS光谱;(b)Mo3d XPS光谱;(c)实验的EXAFS R空间(线性曲线)和拟合数据(散点曲线);(d)对应于曲线c的k空间的WT。
XPS分析表明,具有强界面的Au-MoO2和通过物理混合物获得的Au/MoO2均具有Au特征峰。将在84.2和87.8 eV的结合能分配给两个与纯金属Au0一致的自旋轨道分裂Au 4f7/2和Au 4f5/2。但是,Au/MoO2样品的Au 4f图拟合显示出在86.4和89.5 eV处有其他峰。两种物质的结合能(ΔBE)的差异约为2.0 eV,表明氧化态可能主要由Au1+组成(图2a)。Mo3d的XPS光谱表明,所有样品均具有Mo4+和Mo5+氧化态。与原始的MoO2相比,在更高的结合能值(232.6和235.9 eV)处观察到Mo6+氧化态的其他弱峰。 Mo6+物种的形成可能是由于低价的Mo物种通过将电子捐赠给Au3+而氧化的结果。这暗示Mo6+原子的位置可能是MoO2表面缺陷的位置,这是金纳米颗粒的核,其中可以存在Au-MoOX键(图2b)。
分析了Au L3-edge EXAFS数据,以解释金元素的局部配位环境。图2c是Au箔,Au/MoO2和Au-MoO2的原子径向分布函数(RDF)的曲线。在2.0-3.5Å区域出现的峰代表了Au原子的第一个壳的路径,其强度与平均Au-Au CNs相关。 Au/MoO2的第一个Au壳峰与Au箔上的壳峰非常相似,Au-Au键的CNs和R分别为10.8和2.85 Å,与大尺寸纳米粒子一致。Au-MoO2的第一个Au壳峰出现在2.82 Å,CNs为5.9,较短的原子间距离暗示将存在一些不饱和的Au物种。另一个峰出现在1.98Å区域,可能是来自Au氧化物相,通过与Au-O壳的单层拟合,CNs为0.7。此结果也与XPS数据一致。这表明Au-MoO2在Au物种的局部配位结构上有很大的不同。这种不饱和的Au物种将源自金纳米颗粒和MoO2载体的氧原子之间的强耦合。 R空间的峰值信息通过k空间振荡(k2加权)的小波变换扩展。 WT图表明,Au箔和Au/MoO2在R〜2.5Å,k〜8.0Å-1处具有最大信号,属于Au-Au路径的振荡。相反,在Au-MoO2样品中,Au-Au路径的最大信号在k〜10.0Å-1处移到更高的波数,等高线的脊在两个位置扩展到更短的原子距离(R〜2.0Å)( k〜5.0Å-1和k〜11.0Å-1)(图2d)。这种演变表明,R空间中1.98Å处的峰可归因于两个部分配位物种氧元素和较重的钼元素的贡献。信号强度弱意味着它们的配位主要存在于金纳米颗粒与MoO2的结晶相紧密结合的结区。该EXAFS证据进一步揭示了XPS表征结果,该结果表明Au1+物种是在Au-MoO2上确定的。较短的原子间距表明Au物种具有几个不饱和的Au原子,低配位的Au原子将与金属氧化物上的O原子强烈结合,这可能会降低其他中间物种的吸附能。
2. Au-MoO2纳米片的光学性能表征
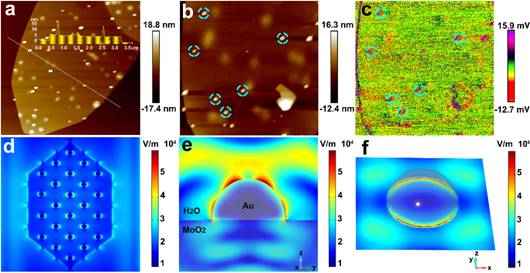
图3 (a)Au-MoO2纳米片的AFM形貌;(b,c)Au-MoO2的形貌和表面电势图像;(d)电磁场分布的二维图;(e,f)Au-MoO2纳米片上的单个颗粒的场分布。
某些特定的金纳米结构具有局部表面等离激元效应,可以大大提高光的能量密度和局部电场的强度。在球体的表面上,具有最大局部场的位置通常是沿着穿过球体中心的轴的两个点,在该点上,感应偶极子和外部场在同一方向上的叠加。在典型的金-半导体混合系统中,等离子体场主要集中在金属和介电基体之间的界面上,同时也是许多催化反应的活性位点。在这种情况下,我们假设局部场强增强主要分布在Au-MoO2的两个不同组分的界面区域。为了研究等离子体场的位置,使用KPFM测量界面区域的接触电势差(CPD)。结果表明,观察到大小为80-100 nm的Au纳米颗粒均匀分布在MoO2纳米片上(图3a),在激光辐射下CPD的高强度区域主要分布在AuNPs周围有环形区域(图3b,c)。该区域的CPD比MoO2衬底(平均3 mV)高21 mV。因此,LSPR产生的电磁场主要位于Au-MoO2结合区域。有限元模拟计算了电磁场分布(图3d),与MoO2衬底相比,Au纳米球周围的结合区具有更强的局域场(Ef/E0> 3500)(图3e,f)。为了进一步证实等离激元效应,计算了不同尺寸的Au纳米颗粒场强分布,较大的颗粒会导致共振变宽,场强急剧下降。相应的紫外可见吸收光谱进一步证实了FEM结果。这主要是由于辐射损失增加和共振红移所引起的,即尺寸效应。此外,界面处的正电场将有助于电子在电催化过程中作为活性位点积累。通过施加负电场,可以提高界面层中金属的费米能级,并将其转移到半导体的导带中以形成简并状态,从而促进半导体向金属行为的转变,从而防止它们发生转变。来自肖特基势垒。
3. Au-MoO2纳米片的电化学析氢性能
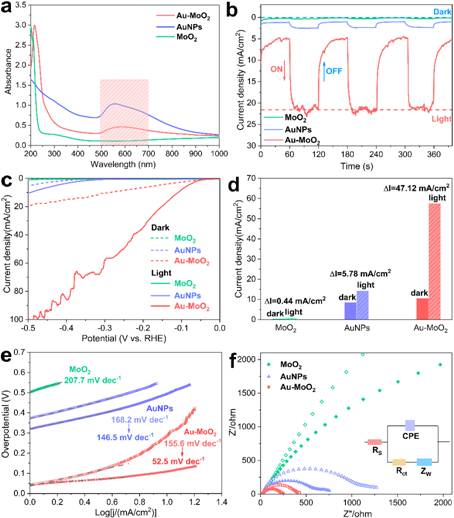
图4 MoO2,AuNPs和Au-MoO2样品的光电化学性能比较:(a)UV-Vis吸收光谱;(b)在-300 mV时的电流时间曲线;(c)极化曲线,(d)在LSV曲线中在-300 mV处的电流密度对比;(e)Tafel斜率;(f)EIS谱;在0.5 M pH 7.5 PBS溶液中进行测试,将电势校准到RHE。
在0.5 M PBS电解质中以5 mV/s的扫描速率对Au-MoO2的等离子体增强HER性能进行了表征。Au-MoO2的吸收光谱表明,与原始MoO2纳米片相比,新的共振吸收带出现在500 nm至700 nm的范围内(图4a)。当入射光子能量低于MoO2带隙时,光电流强度将主要归因于Au的等离激元效应,电流密度表明,激光照射下,与MoO2(2.5 mA/cm2)和AuNPs(0.32 mA/cm2)相比,Au-MoO2电极的最大光电流最大为21.5 mA/cm2(图4b)。在不同的激发光源(包括406 nm,532 nm,650 nm和808 nm)下测试了上述Au-MoO2电极的光电流响应,这也证实光电流主要来源于等离激元效应。同样,电化学响应的增强显示出对Au纳米颗粒大小的重要依赖性,这与电磁场仿真结果一致。
图4c中的LSV表明,所有催化剂在没有光照的情况下都表现出弱的活性,在10 mA/cm2的电流密度下,Au-MoO2的过电势为257 mV。当电极在光照下时,Au-MoO2的电流密度显著增加,与未光照相比,Au-MoO2的光电流增加了大约五倍,电流密度变化ΔI= 47.2 mA/cm2,大于MoO2和AuNPs的电流密度变化。Au-MoO2的过电势显着降低,过电势变化Δη= 176 mV,但光照后Au-MoO2的起始电位几乎没有变化。Au-MoO2的Tafel斜率减小至52.5 mV/dec,远小于图4e中未照明时测得的Tafel斜率(155.6 mV/dec),这表明HER的决速步骤倾向从Heyrovsky机理转变为塔菲尔机理。EIS分析结果表明,当存在光照时,Au-MoO2的电荷转移电阻从361.6 Ω/cm2降低到205.7 Ω/cm2,与HER一致性能(图4f)。
4. Au-MoO2纳米片的电子结构特征
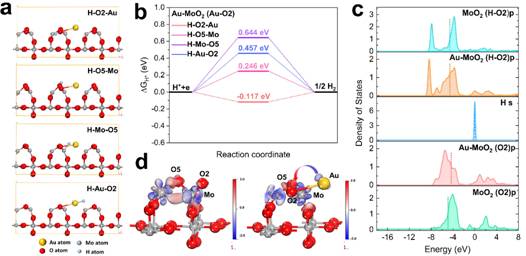
图5 (a)Au-MoO2(010)界面处的Au原子配位结构;(b)在Au,Mo和O的各个位置上H *吸附的吉布斯自由能(ΔGH*),随氢吸收的差分电荷密度分布;(c)氧元素p轨道的PDOS,图中的虚拟垂直线代表计算出的费米能级;(d)MoO2和Au-MoO2上的电荷密度差分。
研究Au-O键对MoO2电子结构和氢吸附能的影响。根据晶相和EXAFS数据,MoO2纳米片的(011)表面被重建和优化。为了更清楚地了解Au-O区域发生的情况,将Au-O-Mo界面定义为MoO2晶面中的O原子与Au原子的通过共价连接,研究两种氧原子(O2,O5)的组合方式。考察了H在Au-MoO2(Au-O2)和Au-MoO2(Au-O5)上的吸附能变化ΔGH*(图5a)。计算结果表明,Au-O2(MoO2)模式具有更好的能量匹配。此外,Au-O2(MoO2)模型中不同位点的ΔGH*分别为0.457 eV(H-Au),-0.117 eV(H-O2),0.246 eV(H-O5)和0.644(H-Mo)(图5b)。结果表明,O2原子是H*原子吸附/解吸过程的最佳途径。
为了进一步了解Au-O-Mo异结构界面上催化活性位点及Au-O键的协同效应,计算了Au-MoO2(Au-O2),Au-MoO2(Au-O5)和MoO2的电子结构。 Au-MoO2(Au-O2)和H吸附的Au-MoO2(Au-O2)中各元素的s,p,d轨道的PDOS表明,O的p轨道由于Au的d轨道的贡献而增加,并且更接近于氢的轨道(图5c)。电荷密度差分表明,电荷积累区域在O2原子附近(红色),而缺电子区域分布在Au原子附近(蓝色)。因此,可以认为主要发生从金到氧原子的电荷转移(图5d)。以上结果表明,通过Au-O键的强耦合相互作用显着加速HER动力学过程。
总结与展望
(1) 通过光辅助沉积构建了 Au-MoO2纳米片的强耦合异质结构;
(2) EXAFS 证实了 Au-MoO2 异质结构界面通过 Au-O 键的紧密耦合;
(3) 表面等离激元的强电磁场通过 Au-O 键诱导表面电荷的聚集。
(4) Au-O键中Op轨道的显著提升降低了氢吸附能。
文章链接
Zhen Liu#, Di Jiang#, Linjing Yang, Jiayuan Yu, Xiao Li, Xiaoyan Liu, Lili Zhao, Xiao Li Zhang, Feng Han, Weijia Zhou*, Hong Liu*, Plasmon-Enhanced Hydrogen Evolution Reaction Kinetics through the Strong Coupling of Au-O Bond on Au-MoO2 Heterostructure Nanosheets, Nano Energy, 2021, 106302, https://doi.org/10.1016/j.nanoen.2021.106302.
第一作者介绍
刘震,山东大学博士,现7321com必赢讲师、博士后。主要研究方向为敏感光电材料在能源催化及分析传感器件的应用。在Nanno energy,Nano-Micro Letters,Biosensors and Bioelectronic,Sci. Rep.等发表多篇论文。主持山东省自然科学青年基金,参与重大科学仪器专项,重大基础研究基金子课题等项目。
 姜迪,硕士研究生,2018年毕业于烟台大学,获学士学位。同年进入BWIN必赢官网前沿交叉学科研究院,加入周伟家教授领导的能源催化材料与器件团队,研究方向为能量转换和存储的纳米材料设计合成及应用,包括电催化,光催化水分解,新型催化材料和储能器件。
姜迪,硕士研究生,2018年毕业于烟台大学,获学士学位。同年进入BWIN必赢官网前沿交叉学科研究院,加入周伟家教授领导的能源催化材料与器件团队,研究方向为能量转换和存储的纳米材料设计合成及应用,包括电催化,光催化水分解,新型催化材料和储能器件。
通讯作者介绍
 刘宏教授,山东大学教授,博士生导师,国家杰出青年科学基金获得者。主要研究方向:纳米材料、生物材料,功能晶体材料、功能陶瓷材料。主持和参加了国家863、973和国家自然科学基金重大项目等10余项,2009年获得国家杰出青年基金项目资助。在包括Nano Letters,J. Am. Chem.Soc,Adv. Mater. Tissue Engineering等SCI学术期刊上发表文章100余篇,引用次数2700余次;申请发明专利20项,授权12项。
刘宏教授,山东大学教授,博士生导师,国家杰出青年科学基金获得者。主要研究方向:纳米材料、生物材料,功能晶体材料、功能陶瓷材料。主持和参加了国家863、973和国家自然科学基金重大项目等10余项,2009年获得国家杰出青年基金项目资助。在包括Nano Letters,J. Am. Chem.Soc,Adv. Mater. Tissue Engineering等SCI学术期刊上发表文章100余篇,引用次数2700余次;申请发明专利20项,授权12项。
 周伟家教授,7321com必赢教授、博士生导师,国家优秀青年基金获得者,入选“泰山学者青年专家计划”,“省优秀青年基金”和“省杰出青年基金”获得者。主要从事纳米材料与技术在电催化、氢能源和微纳器件等领域的研究,主持国家自然科学基金等省部级项目10项。在Energy Environ. Sci.、Angew. Chem. Int. Ed.、ACS Nano等期刊发表SCI收录论文70余篇,被他引6126次,2018年“全球高被引科学家”(交叉学科);2019年山东省自然科学奖一等奖(第三位);授权发明专利6项。
周伟家教授,7321com必赢教授、博士生导师,国家优秀青年基金获得者,入选“泰山学者青年专家计划”,“省优秀青年基金”和“省杰出青年基金”获得者。主要从事纳米材料与技术在电催化、氢能源和微纳器件等领域的研究,主持国家自然科学基金等省部级项目10项。在Energy Environ. Sci.、Angew. Chem. Int. Ed.、ACS Nano等期刊发表SCI收录论文70余篇,被他引6126次,2018年“全球高被引科学家”(交叉学科);2019年山东省自然科学奖一等奖(第三位);授权发明专利6项。
新能源材料与传感器件团队介绍

新能源材料与传感器件团队依托7321com必赢、山东大学晶体材料国家重点实验室。综合利用微纳加工、激光合成和电化学三大技术,在能源与传感两大方向开展应用基础研究。能源方向:氢循环、碳循环和氮循环,与激光、环境、物理信号调制和能源器件系统相关研究;传感方向:电化学、拉曼和荧光检测结合微流控技术,进行细菌、外泌体和分子等生物传感检测相关材料设计与器件构建研究。目前团队有教授2人、副教授1人,讲师3人,博士后2人,博士生4人,硕士生17人。
